cootes@icrf.icnet.uk, stephen@cs.york.ac.uk, sternber@icrf.icnet.uk
Abstract.
It remains unclear what principles underlie a protein sequence/structure adopting a given fold. Local properties such as the arrangement of secondary structure elements adjacent in sequence or global properties such as the total number of secondary structure elements may act as a constraint on the type of fold that a protein can adopt. Such constraints might be considered "signatures" of a given fold and their identification would be useful for the classification of protein structure. Inductive Logic Programming (ILP) has been applied to the problem of automatic identification of structural signatures. The signatures generated by ILP can then be both readily interpreted by a protein structure expert and tested for their accuracy.A previous application of ILP to this problem indicated that large insertions/deletions in proteins are an obstacle to learning rules that effectively discriminate between positive and negative examples of a given fold. Here, we apply an ILP learning scheme that reduces this problem by employing the structural superposition of protein domains with similar folds. This was done in three basic steps. Firstly, a multiple alignment of domains was generated for each type of fold studied. Secondly, the alignment was used to determine the secondary structure elements in each of those domains that can be considered equivalent to one another (the "core" elements of that fold). Thirdly, an ILP learning experiment was conducted to learn rules defining a fold in terms of those core elements.
The relationship between a proteins sequence and its structure and function is complex and, as yet, not fully understood. To a large extent, the current understanding of protein sequence/structure/function has come from careful manual examination. However, the recent explosion in the amount of sequence data from genome projects and the ever increasing numbers of protein structures has highlighted the need for automatic approaches to the analysis of biological problems. One such problem is the identification of the key structural features that define a given protein fold. A previous study [1] that applied Inductive Logic Programming (ILP) [2] to the automatic identification of structural signatures in protein folds found that large insertions and deletions in a protein structure proved to be a major obstacle. This was because the structurally equivalent portions of proteins with the same overall fold could not easily be identified. In this study, we applied a multiple structure alignment technique to identify equivalent sub-structures in proteins with the same fold. ILP was then applied to learn the principles governing that fold in terms of those core sub-structures.
A protein's fold can be defined and classified in terms of the sequential and spatial arrangements of their regular sub-structures (or secondary structure elements):
a-helices and b-strands. There are several fold classification techniques that have already been developed using manual (SCOP [3]), semi-automatic (CATH [4]) and fully automatic techniques (FSSP [5]). Despite the large overlap of these classifications the differences between them are quite significant [6]. The gap between the human expert's understanding of protein sequence/structure and current automated procedures is probably best highlighted by the results of the CASP and CAFASP blind trials [7]. In these trials, human experts and automated servers were asked to predict a protein's structure from its sequence alone. Those methods that employed some level of human intervention outperformed fully automated techniques. This is largely because the human expert has the advantage of drawing on the vast amount of background knowledge collected over years of research that is not normally incorporated into automatic approaches. This could include knowledge of evolutionary relationships, biochemical principles or structural features that are important for a given fold. Such knowledge would allow an expert to screen predictions for those that violate principles already known to them and eliminate them for consideration.Although the intervention of a human expert may improve fold classification or prediction, such intervention is always subjective. Furthermore, knowledge of protein structural principles only extends beyond a small number of fold types for a few protein experts. Hence, it would be desirable to develop a fully automated method by which expert-like structural principles could be derived in an objective manner for all types of protein structure in "fold space". One such method is ILP, a form of machine learning, that can derive rules from examples and background knowledge. ILP has been applied previously to several probems in structural molecular biology [8-12]. In fact, ILP has also been previouly been applied to the discovery of protein structural principles [1]. In that study, significant local features of several folds were found, such as a short loop between the first
a-helix and the following b-strand in proteins with a Rossmann fold, known to be part of a functional binding site. However, important global features of folds such as the topology of b-sheets (the order in which b-strands align with each other in space) and a-helix spatial packing arrangements have proved elusive to learning with ILP. Some folds with well-known global features (e.g. TIM barrels) failed to yield any rules at all. This is because of the large number of exceptions in domains with the same fold. Typically, a domain can have a segment inserted into its structure such that a secondary structure element that is still structurally equivalent to an element in another domain with the same fold can occur much further ahead in sequence and not be recognised as being equivalent. However, there are some standard tools available (such as SSAP [13]) by which a pair of structure can be compared and aligned.In this study, we build multiple structure alignments of all domains with the same fold from pairwise alignments calculated with the SSAP program. Obtaining a reliable multiple structure alignment is typically quite difficult [14] but this enables the identification of structurally equivalent (core) secondary structures in those domains and the elimination of inserted structures (non-core) that inhibit the learning of global structural features. From these core secondary structure elements rules were learnt for several SCOP fold classifications and tested for their accuracy.
The SCOP database is constructed manually and is based on the knowledge of protein expert A. Murzin, taking into account evolutionary relationships between protein sequence, structure and function. This classification scheme offers the advantage that each fold classification has a brief text description of the principles on which each fold type is categorised so that the rules learnt can also be compared to human expert knowledge.
The set of protein domains used for each fold category were obtained from the SCOP database [3], release number 1.50. For learning rules, one domain from each protein/species category within the four main fold classes (all alpha, all beta, alpha/beta and alpha+beta) were selected using the ASTRAL [15] database. This included some related domains from the same SCOP family but this was found to improve both the multiple structure alignments and the quality of the rules learnt as determined by our protein expert (M. Sternberg). However, for the purposes of testing, one domain per SCOP family was selected in order to eliminate redundancy.
In order to define the core structural elements for each domain within a given fold category, a multiple structure alignment of those domains was performed. Multiple structure alignments indicate which residue positions in the aligned domains that can be considered structurally equivalent and eliminate the variable regions in the structures from consideration.
For the purposes of this work, a technique was employed whereby multiple alignments were constructed by clustering pairwise alignments of domains with similar folds. Such a method tends to neglect global features of the multiple alignment but is fast to calculate. Pairwise alignments were generated for each possible pair of domains using the SSAP program [13]. The pairwise alignments were then clustered with respect to the structural similarity of the aligned domain pairs (given by their pairwise RMSD, which decreases with increasing structural similarity), in a similar manner to that in a previous publication [16], to give the final multiple structure alignment.
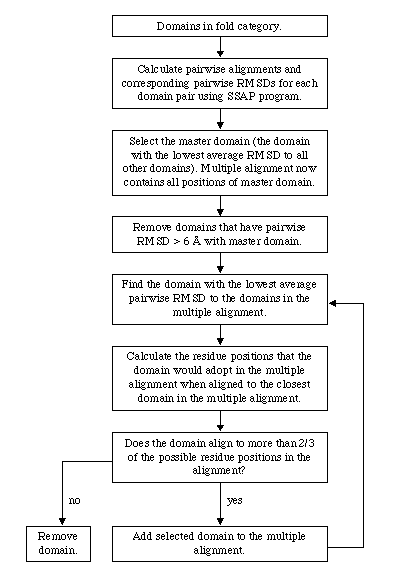
The clustering process is shown schematically in Figure 1 and proceeded as follows: Firstly, a master domain was selected by finding the domain with the lowest average RMSD to all other domains in that fold category. The master domain then acted as a seed for the subsequent alignment of the remaining domains. To eliminate outliers, any domains that had 6 Å RMSD with the master domain were firstly eliminated from further consideration. Then, the domain with the lowest RMSD to the master domain was then aligned to that domain so that the current alignment then contained two domains. The domain with the lowest average RMSD to the domains in the current alignment was then aligned to the closest domain in the current alignment. This process continued iteratively until all domains in that fold category have been considered. In order to avoid corrupting the multiple alignment with misaligned pairwise alignments, domains that align to less than 2/3 of the residue positions in the current alignment at any given step were eliminated from consideration. For most fold categories, only a few domains were eliminated in this way.
An example of a multiple structure alignment in shown in Figure 2.
The multiple structure alignment indicates which residues in each structure can be considered structurally equivalent. However, to learn rules for protein structure in terms of secondary structure elements (blocks of residues with an alpha-helical or beta-strand structure) the elements that can be deemed equivalent have to be identified. To do this, a simple matching scheme was employed to match secondary structures units in different domains based on the level of overlap of those units in the multiple alignment calculated previously (shown schematically in Figure 3).
The secondary structure for each protein in the multiple structure alignment was determined using the PROMOTIF [17] program. Firstly, those secondary structure elements that have less than half of their constituent residues aligned were removed from consideration. Then, all pairwise "matches" between secondary structure elements in each pair of aligned proteins were determined. A "match" was deemed to have occurred between two secondary structure elements from different proteins if each was the largest overlapping element of the other in their respective proteins. Secondary structure elements were then grouped into "maximally matched" groups (ie. each member of the group is "matched" with every other member of that group). Surprisingly, in some cases this was enough to find equivalent secondary structures in every protein. However, a more relaxed matching scheme is required to find some of the less easily identifiable core elements. Therefore, groups of "sub-maximally matched" elements were identified by breaking up the smallest maximally matched group and redistributing its individual member elements to the largest group for which that element matches more than 1/2 of the constituent members of that group. If no such group could be found then the element was eliminated from further consideration. This process continued iteratively until the only remaining groups contained elements in more than 2/3 of the aligned domains. The remaining elements were deemed to be core elements and equivalent to the other member elements in the same group. Each core group is labelled according to its position in the sequence (i.e. the first group is labelled "a", the second "b" and so on).
Once the core elements for a protein structure have be defined, the background knowledge containing the structural information for that example can be determined in terms of those core elements. The predicates describing the attributes of, and relationships between, core elements that were considered here and their descriptions are listed in Table 1. All of the structural information required for determining the background knowledge was taken from the output of the PROMOTIF [17] program for that example.
Rules were learnt for each SCOP fold category in which protein domains from more than 5 sequence families could be aligned (shown in Table 2) using the Progol-4.4 ILP system. Progol is described in more detail elsewhere [18,19], below is a brief description of the algorithm.
Positive examples were taken from the fold category of interest and the negative examples were taken from all other fold categories in the same SCOP main fold class (all
a, all b, a/ b or a+ b). Thus, the negative examples were selected only from the most similar folds to the positive examples on the premise that it is more difficult to discriminate against these folds.Progol then proceeds to learn a rule by selecting a positive example and collecting all related background information. It then builds steadily more specific rules from that information until its measure of compression is maximised. The measure of compression used f is:
f = p - n - c
where p is the number of positive examples covered by the rule, n is the number of negative examples covered and c is the length of the rule. The parameter c ensures that for rules with equal coverage of positive and negative examples the shorter one is favoured (i.e. the one that obeys the principle of parsimony). Once an optimal rule is found, the positive examples covered by that rule are removed and the process begins again. This continues until no positive examples remain.
The maximum number of nodes (or hypotheses) tested allowed for an individual search was set to 1000. The noise parameter controlled the number of negative examples that a rule was allowed to cover. The level of noise allowed was 20% (i.e. up to 20% of examples covered by a rule could be false positives).
5-fold cross-validation testing was performed for rules learnt for each fold category considered.
Rules were learnt for each SCOP fold type in the four main classes (all-
a, all-b, a/b and a+b) for which representative domains for more than 5 sequence families (and hence more than 5 test examples could be aligned) (Table 2). 5-fold cross-validation tests were conducted for each fold type to determine the accuracy, precision and recall of those rules found. The overall accuracy for the folds considered here is 98% (a random result would be 91%) which is significant according to a c2 test, giving a probability p < 0.0001that the result could have occurred by chance. This result is dominated by the testing of negative examples but the overall precision and recall (85% and 63% respectively) is reasonably high.The overall results for each main fold class are all significant, but those folds of the all-b class have a much lower recall (34%) than the remaining classes. Several folds in this class do not find any rules at all. This appears to be due to problems with the alignments of b-sheets, although this hasn't presented as much of a problem with the a/b or a+b main fold classes. Indeed, the latter two classes appear to have better overall recall and precision than either the all-a or all-b classes for the fold types studied here. This contrasts with the results of previous ILP learning experiments without the aid of multiple alignments [1].
The composition of the rules that were learnt for all folds as given in the previous section are shown in Table 3.
The rules learnt for the fold types here appear to be dominated by sheet topology overall. Combining the occurrence of all sheet_top_X predicates, where X is the number of
b-strands in the sheet, reveals that 52% of rules learnt contain such a predicate. Learning the topology of b-sheets in a fold is a difficult task. However, once the core elements have been extracted from the folds via a multiple alignment, sheet topology in terms of those elements can be learnt more easily. Other prominent predicates proved to be those describing the angles between contacting helices, general contacts between secondary structure elements and the presence of glycine or proline. Descriptors that proved to be quite prevalent in rules learnt previously for folds [1], such as the length of loops, did not occur at all in rules learnt in this study.Perhaps the most interesting difference between this study and previous work using ILP to discover structural signatures is the ability of this method to capture the global features of folds familiar to human experts. In this section, rules are presented for three important folds and compared to rules learnt previously with ILP without structural alignments [1] and the text descriptions of those folds that have been provided on the SCOP [3] website. The descriptions provided by SCOP are preliminary, and do not represent the sum total of expert-knowledge of the fold, but do give a general expert guide to the general features of the fold. Rules learnt using Progol are output in terms of clauses consisting of the combinations of the types of predicate listed in Table 1, but for clarity and ease of comparison, the rules so learnt have been interpreted into english statements similar to that of the SCOP descriptions. The rules given for Immunoglobulin-like beta-sandwich (SCOP classification 1 002 001), TIM barrels (1 003 001) and Rossmann-like folds (1 003 002) are shown in Table 4.
For the Immunoglobulin fold, the important features of the fold according to the experts who designed SCOP are two
b-sheets, consisting of 7 strands between them, flat against each other in space much like two layers in a sandwich. It also contains a small b-strand motif involving connections between strands in opposite sheets, known as a greek key motif. The previous application of ILP without structural alignments identified one attribute (that Immunoglobulins sometimes have a helix present) and also found a local feature (small loop between the 5th and 6th strands) of the fold. With the use of multiple structure alignments however, a global structure description much closer to that of a human expert is obtained. In one rule, it not only finds that there are 7 strands in two sheets but identifies the topology of each. The second rule gives a partial description of which strands in the sheets come into contact in order to form the tertiary structure (the "sandwich" packing of the two sheets).The previous application of ILP to this problem failed to find a rule for the TIM barrel fold. This was largely due the large number of structural variations in TIM barrels. However, the overall fold and global features of TIM barrels are well known. SCOP describes the fold as having a parallel
b-sheet with 8 strands (n=8) folded around so that the end strands meet each other to form a closed barrel. It also states that the strands in the sheet are ordered 12345678 and gives other properties that are not included in our ILP representation here, such as the degree to which the strands are "staggered" with respect to each other (S=8). In this study, ILP found a rule for the TIM barrel fold in terms of global features, although it did not have the same depth of detail with regard to sheet topology and geometry as the SCOP description. ILP found a rule that described the number of helices in a TIM barrel (between 5 and 9 core a-helices) and the number of strands in the parallel sheet (8) but did not identify the order of the strands in the sheet or identify the sheet as a closed barrel. However, it is known that some TIM barrel domains have barrels that are not entirely "closed" [20] i.e. the sheet is curved in space but the strands on either end of the sheet do not quite meet (this is known as an "open" barrel). Such a structure is not recognised by the representation used here as being a barrel and hence, such a rule was not found by ILP.For the Rossmann-like folds, the method used in this study finds two rules. Firstly, it identifies a Rossmann-like fold as a domain with between 3 and 4
a-helices, with the a-helix at core position "b" having a glycine in both its middle and n-terminal region. This rule has identified two glycines of a known conserved G-X-G-X-X-G sequence motif [21], where G is a glycine and X is any type of amino acid, involved with binding the adenosine of NAD. This is a conserved functional, rather than simply a structural, feature. The previous application of ILP to this problem [1] also located the loop between the 1st strand and the 2nd helix where this conserved region is located. The second rule found in this study for the Rossmann-like fold describes global structural features of the fold. It identifies that the fold has a 6 strand parallel sheet with topology 321456 and also describes two of the helices that are in contact and parallel to one another in space. This is quite similar to the global features given in the SCOP description of the fold. SCOP describes a 6 strand parallel sheet with topology 321456 but does not give structural details of the a-helices except to point out that helices pack on either side of the sheet in 3 layers (a/b/a).This study shows that for those fold types that have a reasonable number of examples, expert-like rules can be learnt in a systematic fashion. Furthermore, given that the core sub-structures of the fold can be reliably identified, the significant global features of folds, such as the topology of
b-sheets or packing of a-helices, can be described. Some of the rules that have been learnt here clearly reflect some of the principles used by the expert who manually constructed the fold classification system from which the rules were learnt. Given the explosion in the number of structures in recent times, constructing such fold classification schemes manually will become increasingly difficult and an automated approach to derive principles of protein structure, such as the one used in this study, will be increasingly necessary.However, the approach to learning structural principles from multiple structure alignments of protein domains used here is currently limited to the well-represented SCOP fold types, as multiple structure alignments used here become far less reliable in defining core structural components when there are only a few domains as examples. As the majority of fold types defined by the SCOP classification have very few domain examples, the method used here may not prove to be as useful when applied to all possible fold categories. An automated method that could extract structural principles from less well represented folds would be far more useful generally. Human experts have a better understanding of the well-represented folds and an automated method may simply give them structural features that they already know for these folds. However, human expertise does not generally extend to many of the less-well represented folds and automated methods of knowledge discovery could yield useful insight in these cases.
Given the low level of data for most fold types, directly application of multiple structure alignment may be difficult and inaccurate in determining the core sub-structures. However, it may be possible to learn principles that can predict which secondary structure elements are core and which are non-core. Intuitively, one might suspect that secondary structure elements that are quite small, on the ends of
b-sheets or do not make many contacts with other parts of the structure may be more variable than those that are not. Such elements, with fewer physical constraints, may be more likely to be non-core. ILP may be able to learn such rules for core and non-core elements from those examples here whose multiple alignments are more reliable. If such rules proved to be physically and biologically sensible, they could be transferred to those folds with fewer examples to predict the core elements of a fold. Then, ILP could again be used to derive structural principles from the predicted core elements in a similar way to that used in this study.Apart from extending this method to folds with fewer examples, other improvements could be made to this method. The representation used here (Table 1) does not include sequence motifs known to be associated with particular functions, such as those collated in the PROSITE [22] database. The inclusion of such motifs could give further insight into the relationships between sequence/structure/function and assist in fold classification and prediction. Currently, the only sequence properties that are represented here are the presence of glycines and prolines in secondary structure elements.
This study shows that ILP can learn global structure principles for fold after identifying the core structural elements via a multiple alignment. The rules learned for well-represented folds reflect principles known to protein experts.
This work was supported by a BBSRC grant.
[1] Turcotte, M., Muggleton, S.H. and Sternberg, M.J. (2001) Journal of Molecular Biology, 306, 591-605.
[2] Muggleton, S.H. and Raedt, L.D. (1994) Journal of Logic Programming, 19/20, 629-679.
[3] LoConte, L., Ailey, B., Hubbard, T.J.P., Brenner, S.E., Murzin, A.G. and Chothia, C. (2000) Nucleic Acids Research, 28, 257-259.
[4] Pearl, F.M.G., Lee, D., Bray, J.E., Sillitoe, I., Todd, A.E., Harrison, A.P., Thornton, J.M. and Orengo, C.A. (2000) Nucleic Acids Research, 28, 277-282.
[5] Holm, L. and Sander, C. (1998) Nucleic Acids Research, 26, 316-319.
[6] Hadley, C, Jones, D.T. (1999) Structure Fold. Des., 7,1099-112.
[7] Moult, J., Hubbard, T., Fidelis, K. and Pedersen, J.T. (1999) Proteins, 37 (S3), 2-6.
[8] Muggleton, S.H., King, R.D. and Sternberg, M.J. (1992) Protein Engineering, 5, 647-657.
[9] King, R.D., Muggleton, S.H., Lewis,R.A. and Sternberg, M.J. (1992) Proc. Nat. Acad. Sci. USA, 89, 11322-6.
[10] King, R.D., Clark, D.A., Shirazi, J. and Sternberg, M.J. (1994) Protein Engineering, 7, 1295-1303.
[11] Hirst, J.D., King, R.D., and Sternberg, M.J. (1994) Journal of Computer-aided Molecular Design, 8, 405-20.
[12] King, R.D., Muggleton, S.H., Srinivasan, A. and Sternberg, M.J. (1996) Proc. Nat. Acad. Sci. USA, 93, 438-42.
[13] Taylor, W .R. and Orengo, C.A. (1989) Journal of Molecular Biology, 208,1-22.
[14] Gerstein, M. and Levitt, M. (1998) Protein Science, 7, 445-456.
[15] Brenner, S.E., Koehl, P. and Levitt, M. (2000) Nucleic Acids Research, 28, 254-256.
[16] Kelley, L.A., MacCallum, R.M. and Sternberg, M.J.E. (2000) Journal of Molecular Biology 299, 499-520.
[17] Hutchinson, E.G. and Thornton, J.M. (1996) Protein Science, 212-220.
[18] Muggleton, S.H. (ed.) (1992) Inductive Logic Programming. Academic Press, London.
[19] Muggleton, S.H. (1995) New Generation Computing Journal, 13, 245-286.
[20] Nagano, N., Hutchinson, E.G. and Thornton, J.M. (1999) Protein Science, 8, 2072-84.
[21] Wierenga, R.K., Terpstra, P. and Hol, W.G.J. (1986) Journal of Molecular Biology, 187, 101-107.
[22] Hofmann, K., Bucher, P., Falquet, L. and Bairoch, A. (1999) Nucleic Acids Research, 27, 215-219.
Figure 1.
Flow diagram describing the construction of a multiple structure alignment for a given fold category.
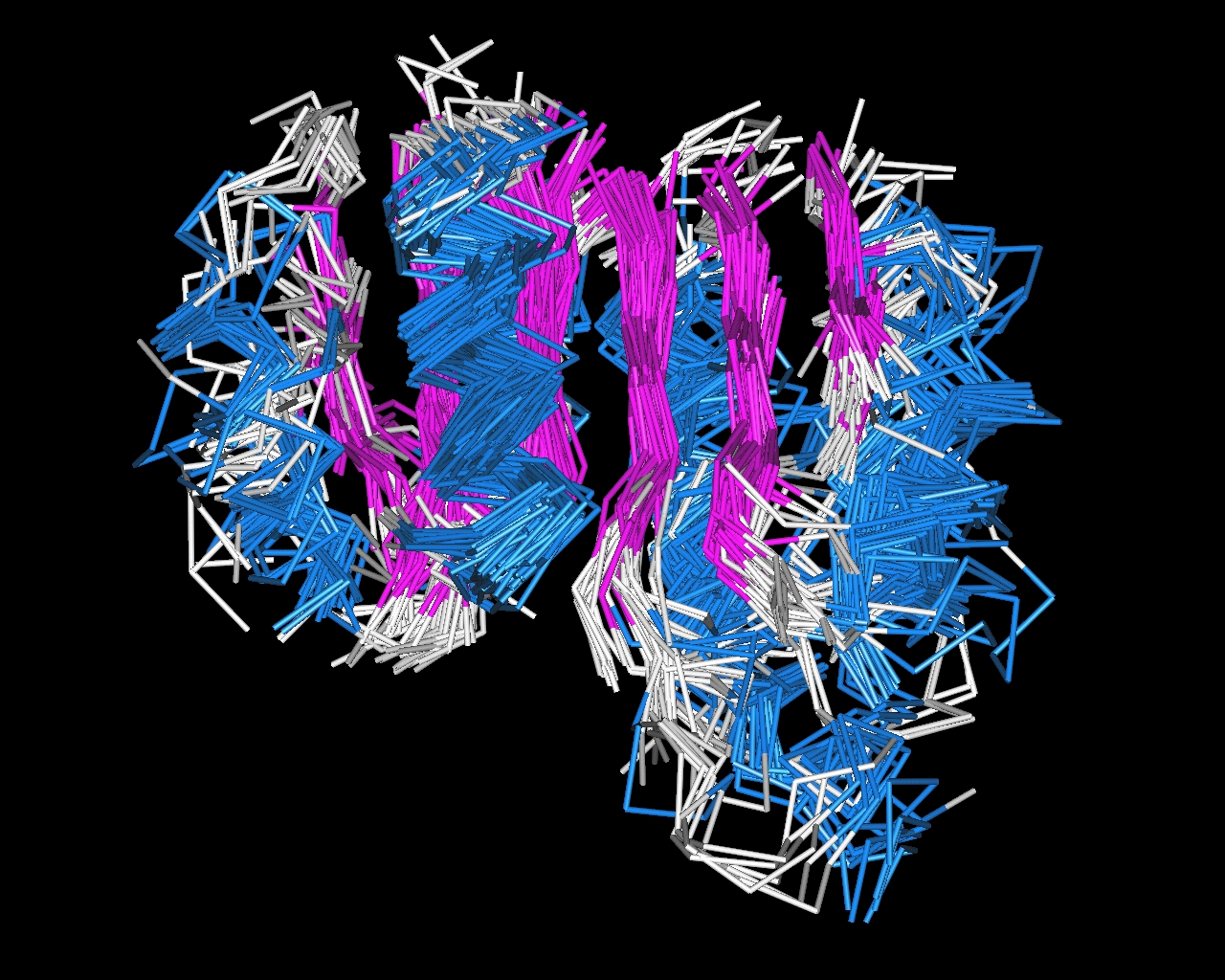
Figure 2. A multiple structure alignment of domains with a Rossmann-like fold. The aligned a-helices are shown in blue and the aligned b-strands are shown in magenta. The six b-strands form a b-sheet.
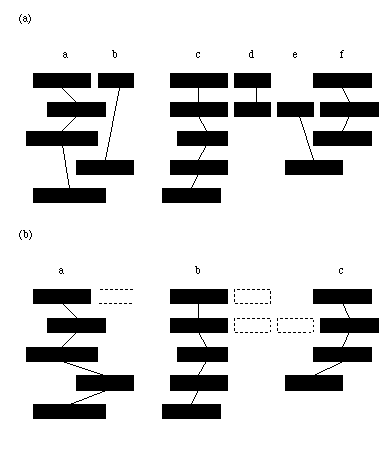
Figure 3. Finding equivalent secondary structure elements in a multiple alignment. Represented are 5 proteins in a multiple structure alignment, aligned so that sequences are running horizontally. Rectangles represent secondary structure elements. Parts of those elements that are vertically above one another are considered to be structurally equivalent. (a) Maximally matched groups of elements are identified (groups are elements connected by lines). (b) Smaller groups are disbanded and their member elements are matched to the larger groups of elements using a less strict matching criterion. Dotted rectangles are the elements deemed to be non-core.
Table 1. The predicates from which rules were learnt. Each predicate is a logical expression in Prolog describing attributes of, or relationships between, elements in a protein domain.
number_helices(Lo =< D =< Hi): The number of helices in domain D.
sheet(D, A Stype): Domain D has a b -sheet A of type Stype, where Stype could be antiparallel, parallel or mixed.
helix(D, B, Htype, Core): Domain D has an helix B at core position Core. B is of type Htype, where Htype can be an a -helix or a 3-10-helix.
strand_position(A, B, N): b -Sheet A has a b -strand B which is the Nth strand in that sheet.
adjacent(B, C): Secondary structure elements B and C are adjacent in sequence.
coil(B, C, N): Elements B and C are adjacent in sequence, separated by a coil of N residues.
contact(B, C): Elements B and C are in contact in space.
antiparallel(B, C): b -strands B and C are antiparallel.
parallel(B, C): b -strands B and C are parallel.
end_strand_distance(A, B, C, Dist): Strands B and C are the end strands of sheet A and are separated by distance Dist in space.
pair(B, C, Bloc, Cloc): Helices B and C are in contact. The parts (N-terminal, C-terminal or middle) of the helices B and C in contact are Bloc and Cloc respectively.
helix_angle(B, C, Angle): Helices B and C are in contact. B and C make angle Angle with each other, where Angle could be antiparallel, parallel or perpendicular.
has_n_strands(A, N): Sheet A has a total of N strands.
barrel(A): Sheet A is a barrel.
bifurcated(A): Sheet A contains a bifurcation.
sheet_top_X(A, N1, N2,…., NX): Sheet A contains X strands, with topology N1N2…..NX (i.e. the N's give the relative sequence order of the strands that are spatially adjacent in the sheet).
contains(B, AA, Loc): Element B contains amino acid AA at location Loc, where AA can be either glycine or proline and Loc can be the N-terminal, C-terminal or middle of the element.
contains(B, AA): As above, but independent of location in the element.
Table 2. Cross-validation results. The cross-validated accuracy is shown for each individual fold category, the four main SCOP fold classes and for all folds combined. The columns give, respectively, the SCOP fold class, the numbers of positive and negative examples, the cross-validated accuracy and error, the accuracy expected given a random guess, the c2 significance, the corresponding probability p, the precision (proportion of positive predictions that are true positives) and the recall (proportion of positive examples that are correctly predicted).
|
Fold |
pos/neg |
Acc. (%) |
Rand. (%) |
c 2 |
p |
Prec. (%) |
Recall (%) |
|
1 001 002 |
7/ 229 |
96 +/- 1 |
96 |
3.40 |
0.0651 |
0 |
0 |
|
1 001 004 |
30/ 206 |
97 +/- 1 |
77 |
178.89 |
0.0000 |
88 |
93 |
|
1 001 025 |
10/ 226 |
96 +/- 1 |
93 |
31.86 |
0.0000 |
50 |
40 |
|
1 001 041 |
9/ 227 |
97 +/- 1 |
93 |
62.07 |
0.0000 |
62 |
56 |
|
1 001 060 |
10/ 226 |
96 +/- 1 |
94 |
26.36 |
0.0000 |
60 |
30 |
|
1 001 110 |
8/ 228 |
97 +/- 1 |
96 |
31.58 |
0.0000 |
100 |
25 |
|
74/1342 |
97 +/- 0 |
91 |
547.66 |
0.0000 |
74 |
57 |
|
|
1 002 001 |
16/ 174 |
96 +/- 1 |
88 |
78.85 |
0.0000 |
100 |
50 |
|
1 002 002 |
7/ 183 |
99 +/- 1 |
94 |
107.82 |
0.0000 |
100 |
71 |
|
1 002 017 |
7/ 183 |
96 +/- 1 |
96 |
nan |
nan |
0 |
0 |
|
1 002 032 |
7/ 183 |
96 +/- 1 |
96 |
6.08 |
0.0137 |
0 |
0 |
|
1 002 038 |
12/ 178 |
97 +/- 1 |
90 |
91.99 |
0.0000 |
100 |
58 |
|
1 002 040 |
6/ 184 |
97 +/- 1 |
97 |
nan |
nan |
0 |
0 |
|
1 002 041 |
7/ 183 |
97 +/- 1 |
95 |
28.97 |
0.0000 |
100 |
29 |
|
1 002 064 |
6/ 184 |
97 +/- 1 |
96 |
3.15 |
0.0758 |
50 |
17 |
|
68/1452 |
97 +/- 0 |
94 |
435.06 |
0.0000 |
92 |
34 |
|
|
1 003 001 |
30/ 181 |
94 +/- 2 |
78 |
111.52 |
0.0000 |
91 |
67 |
|
1 003 002 |
6/ 205 |
99 +/- 1 |
94 |
116.38 |
0.0000 |
83 |
83 |
|
1 003 016 |
15/ 196 |
97 +/- 1 |
88 |
110.39 |
0.0000 |
91 |
67 |
|
1 003 042 |
6/ 205 |
99 +/- 1 |
94 |
116.38 |
0.0000 |
83 |
83 |
|
1 003 064 |
17/ 194 |
98 +/- 1 |
86 |
133.56 |
0.0000 |
93 |
76 |
|
74/ 981 |
97 +/- 0 |
88 |
643.79 |
0.0000 |
90 |
72 |
|
|
1 004 013 |
8/ 255 |
99 +/- 1 |
94 |
171.12 |
0.0000 |
88 |
88 |
|
1 004 014 |
6/ 257 |
100 +/- 0 |
96 |
220.06 |
0.0000 |
100 |
100 |
|
1 004 015 |
7/ 256 |
100 +/- 0 |
94 |
196.70 |
0.0000 |
88 |
100 |
|
1 004 048 |
32/ 231 |
98 +/- 1 |
79 |
218.29 |
0.0000 |
94 |
94 |
|
1 004 076 |
7/ 256 |
98 +/- 1 |
94 |
122.89 |
0.0000 |
67 |
86 |
|
60/1255 |
99 +/- 0 |
91 |
1060.30 |
0.0000 |
89 |
93 |
|
|
Overall |
276/5030 |
98 +/- 0 |
91 |
2743.06 |
0.0000 |
85 |
63 |
Table 3. Composition of the rules learnt. Given are the relative proportion of rules learnt containing at least one of each type of predicate.
|
Predicate |
Percentage of rules containing predicate |
|
|
|
|
helix |
40.74 |
|
sheet |
33.33 |
|
strand_position |
22.22 |
|
helix_angle |
22.22 |
|
contact |
18.52 |
|
sheet_top_4 |
18.52 |
|
contains |
18.52 |
|
sheet_top_5 |
18.52 |
|
pair |
11.11 |
|
sheet_top_3 |
7.41 |
|
parallel |
7.41 |
|
has_n_strands |
7.41 |
|
end_strand_distance |
7.41 |
|
sheet_top_6 |
3.70 |
|
sheet_top_7 |
3.70 |
|
antiparallel |
3.70 |
|
adjacent |
3.70 |
Table 4. Rules learnt for several fold types. Rules learnt using the method used in this study (ILP(new)) are compared to the rules learnt previously with ILP without multiple structure alignment (ILP (old)) and expert-like descriptions of those folds taken from the SCOP database (SCOP). Terms used in the SCOP descriptions are described in section 3.3.
|
SCOP fold class |
Rule type |
Rule |
|
Immunoglobulin (1 002 001) |
SCOP |
sandwich; 7 strands in 2 sheets; greek-key; some members of the fold have additional strands |
|
ILP (old) |
There is at most one helix, the loop between the 5th and 6th strands is three to seven residues long. |
|
|
ILP (new) |
Has antiparallel sheets B and C; B has 3 strands, topology 123; C has 4 strands, topology 2134. OR Has antiparallel sheets B and C; C has 3 strands, topology 123; the 1st and 2nd strands in B and D and E respectively; the 1st and 2nd strands in C are F and G respectively; E and F are in contact; D and G are in contact. |
|
|
TIM barrel (1 003 001) |
SCOP |
contains parallel beta-sheet barrel, closed; n=8, S=8; strand order 12345678; the first six superfamilies have similar phosphate-binding sites |
|
ILP (old) |
no rule found |
|
|
ILP (new) |
Has between 5 and 9 helices;Has a parallel sheet of 8 strands. |
|
|
Rossmann-like (1 003 002) |
SCOP |
core: 3 layers, a/b/a; parallel beta-sheet of 6 strands, order 321456; The nucleotide-binding modes of this and the next two folds/superfamilies (1 003 003 and 1 003 004) are similar |
|
ILP (old) |
The 1st strand is followed by a helix, the two elements are separated by a coil of about one residue.The 6th strand is followed by a helix. |
|
|
ILP (new) |
Has between 3 and 4 helices; Has a-helix B at core position "b"; B contains a glycine in both it's middle and n-terminal regions.OR Has a parallel sheet B of six strands with topology 321456; Has a-helices C and D at core positions "g" and "i" respectively; C and D are in contact and parallel. |